First I like to use a quantitative framework (you can use ranks-based indices as in Williams et al 2011, which has nice properties too). The simplest quantitative index is the forage ratio:
fr <- function(obs, exp){
p_obs <- obs/sum(obs)
p_exp <- exp/sum(exp)
out <- p_obs/p_exp
out
}
But it ranges from 0 to infinity, which makes it hard to interpret.
Second, the most used index is “Electivity” (Ivlev 1961), which is nice because is bounded between -1 and 1. “Jacobs” index is similar, but correcting for item depletion, which do not apply to my question, but see below for completness.
electicity <- function(obs, exp){
p_obs <- obs/sum(obs)
p_exp <- exp/sum(exp)
out <- (p_obs-p_exp)/(p_obs+p_exp)
}
jacobs <- function(obs, exp){
p_obs <- obs/sum(obs)
p_exp <- exp/sum(exp)
out <- (p_obs-p_exp)/(p_obs+p_exp-2*p_obs*p_exp)
out
}
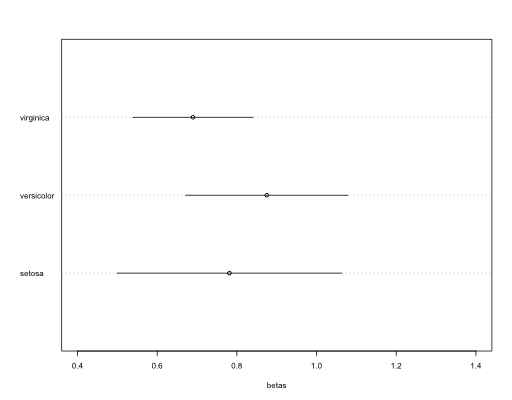
However, those indexes do not tell you if pollinators have general preferences over several items, and which of this items are preferred or not. To solve that we can use a simple chi test. Chi statistics can be used to assess if there is an overall preference. The Chi test Pearson residuals (obs-exp/√exp) estimate the magnitude of preference or avoidance for a given item based on deviation from expected values. Significance can be assessed by building Bonferroni confidence intervals (see Neu et al. 1974 and Beyers and Steinhorst 1984). That way you know which items are preferred or avoided.
chi_pref <- function(obs, exp, alpha = 0.05){
chi <- chisq.test(obs, p = exp, rescale.p = TRUE)
print(chi)
res <- chi$residuals
alpha <- alpha
k <- length(obs)
n <- sum(obs)
p_obs <- obs/n
ak <- alpha/(2*k)
Zak <- abs(qnorm(ak))
low_interval <- p_obs - (Zak*(sqrt(p_obs*(1-p_obs)/n)))
upper_interval <- p_obs + (Zak*(sqrt(p_obs*(1-p_obs)/n)))
p_exp <- exp/sum(exp)
sig <- ifelse(p_exp >= low_interval & p_exp <= upper_interval, "ns", "sig")
plot(c(0,k+1), c(min(low_interval),max(upper_interval)), type = "n",
ylab = "Preference", xlab = "items", las = 1)
arrows(x0 = c(1:k), y0 = low_interval, x1 = c(1:k), y1 = upper_interval, code = 3
,angle = 90)
points(p_exp, col = "red")
out <- data.frame(chi_test_p = rep(chi$p.value, length(res)),
chi_residuals = res, sig = sig)
out
}
And we wrap up all indexes…
preference <- function(obs, exp, alpha = 0.05){
f <- fr(obs, exp)
e <- electicity(obs, exp)
c <- chi_pref(obs, exp, alpha = alpha)
out <- data.frame(exp = exp, obs = obs, fr = f, electicity = e, c)
out
}
It works well for preferences among items with similar availability, and when all are used to some extent. The plot shows expected values in red, and the observed confidence interval in black. If is not overlapping the expected, indicates a significant preference.
x <- preference(obs = c(25, 22,30,40), exp = c(40,12,12,53))

## Chi-squared test for given probabilities
##
## data: obs
## X-squared = 44.15, df = 3, p-value = 1.404e-09
## exp obs fr electicity chi_test_p chi_residuals sig
## 1 40 25 0.6250 -0.2308 1.404e-09 -2.372 sig
## 2 12 22 1.8333 0.2941 1.404e-09 2.887 ns
## 3 12 30 2.5000 0.4286 1.404e-09 5.196 sig
## 4 53 40 0.7547 -0.1398 1.404e-09 -1.786 sig
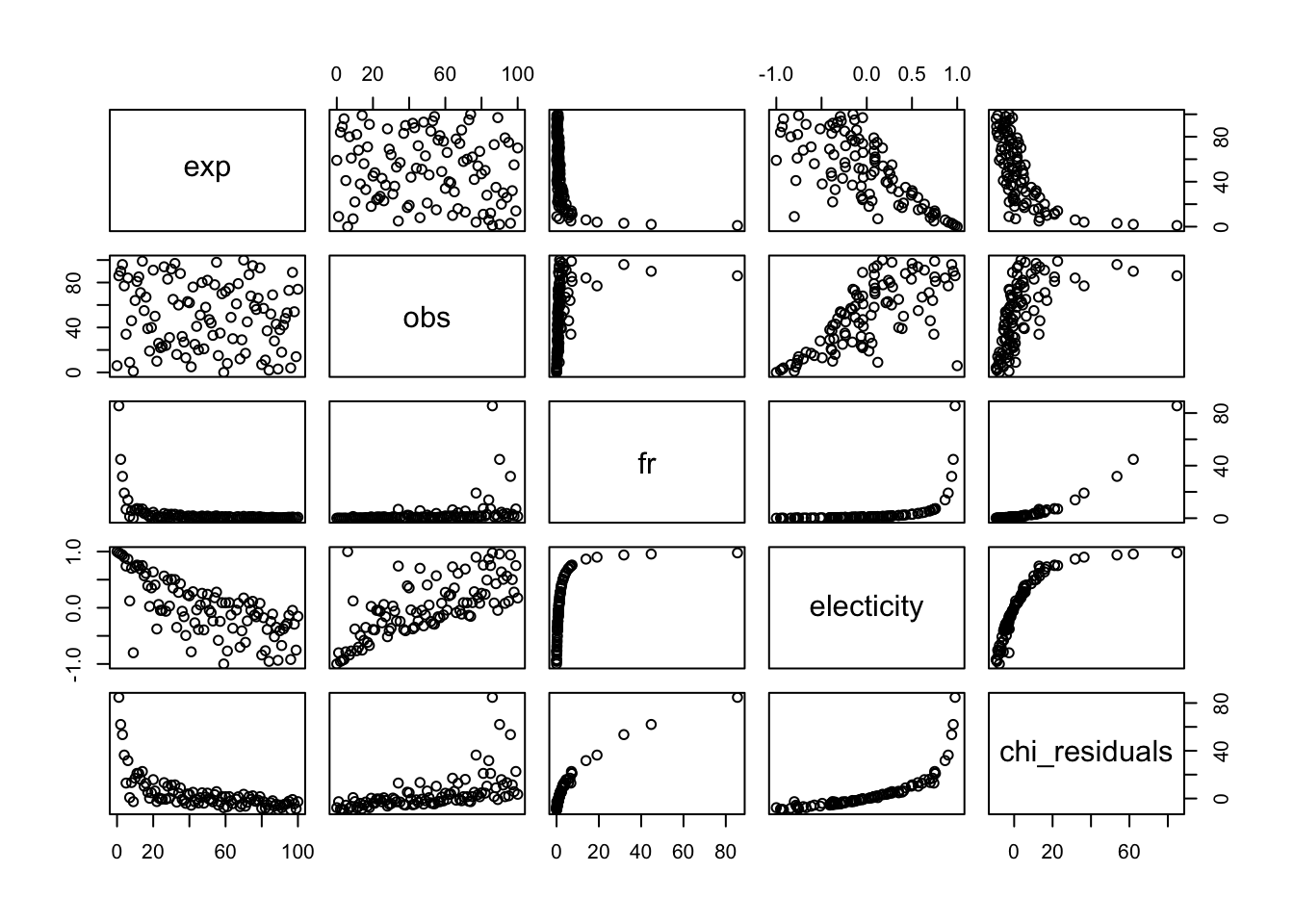
We can see that indices are correlated by simulating lots of random values.
x <- preference(obs = sample(c(0:100),100), exp = sample(c(0:100),100))
## Chi-squared test for given probabilities
##
## data: obs
## X-squared = Inf, df = 99, p-value < 2.2e-16
pairs(x[,c(-5,-7)])

But the indexes do not behave well in two situations. First, when an item is not used, it is significanly un-preferred regardless of its abundance. I don’t like that, because a very rare plant is expected to get 0 visits from a biological point of view. This is an issue when building bonferroni intervals around 0, which are by definition 0, and any value different from 0 appears as significant.
x <- preference(obs = c(0,25,200), exp = c(0.00003,50,100))

## Chi-squared test for given probabilities
##
## data: obs
## X-squared = 50, df = 2, p-value = 1.389e-11
## exp obs fr electicity chi_test_p chi_residuals sig
## 1 3e-05 0 0.0000 -1.0000 1.389e-11 -0.006708 sig
## 2 5e+01 25 0.3333 -0.5000 1.389e-11 -5.773502 sig
## 3 1e+02 200 1.3333 0.1429 1.389e-11 4.082486 sig
The second issue is that if you have noise due to sampling (as always), rare items are more likely to be wrongly assessed than common items.
x <- preference(obs = c(5,50,100), exp = c(5,50,100))

## Chi-squared test for given probabilities
##
## data: obs
## X-squared = 0, df = 2, p-value = 1
## exp obs fr electicity chi_test_p chi_residuals sig
## 1 5 5 1 0 1 0 ns
## 2 50 50 1 0 1 0 ns
## 3 100 100 1 0 1 0 ns
x <- preference(obs = c(1,54,96), exp = c(5,50,100))

## Chi-squared test for given probabilities
##
## data: obs
## X-squared = 3.671, df = 2, p-value = 0.1595
## exp obs fr electicity chi_test_p chi_residuals sig
## 1 5 1 0.2053 -0.659341 0.1595 -1.7539 sig
## 2 50 54 1.1086 0.051508 0.1595 0.7580 ns
## 3 100 96 0.9854 -0.007338 0.1595 -0.1438 ns
As a conclusion, If you have all items represented (not highly uneven distributions) or all used to some extent (not 0 visits) this is a great tool.
Happy to know better options in the comments, if available.
Gist with the code of this post here.